
Case study: Penumbra’s JET 7 Reperfusion Catheter Class 1 Recall
The recent recall of the Penumbra JET™ 7 reperfusion catheter came after 200 adverse events (AEs), including 14 patient deaths. The Class I device is intended to remove blood clots in the brain that cause stroke. The version of the product the FDA approved in 2019 was nearly 20 years in the making across incremental changes to several related products.
Its recall has triggered industry experts – including as reported by RAPS and JAMA – to reiterate long-held concerns about the lack of necessary and effective mechanisms within the post-market surveillance system to identify and address serious patient risks. Although recalls of complex medical devices can never be completely avoided, this recall caught attention because the reported adverse events didn’t seem to be leading to appropriate corrective action. The long history of incremental developments supporting Penumbra’s FDA clearance adds to the ongoing discussion around what should constitute sufficient “clinical evidence” for safety and efficacy of cleared devices. These are important questions. Let’s take a closer look at what the data tells us.
The data shows an immediate onset of reported AEs after commercial launch, but these did not attract much notice..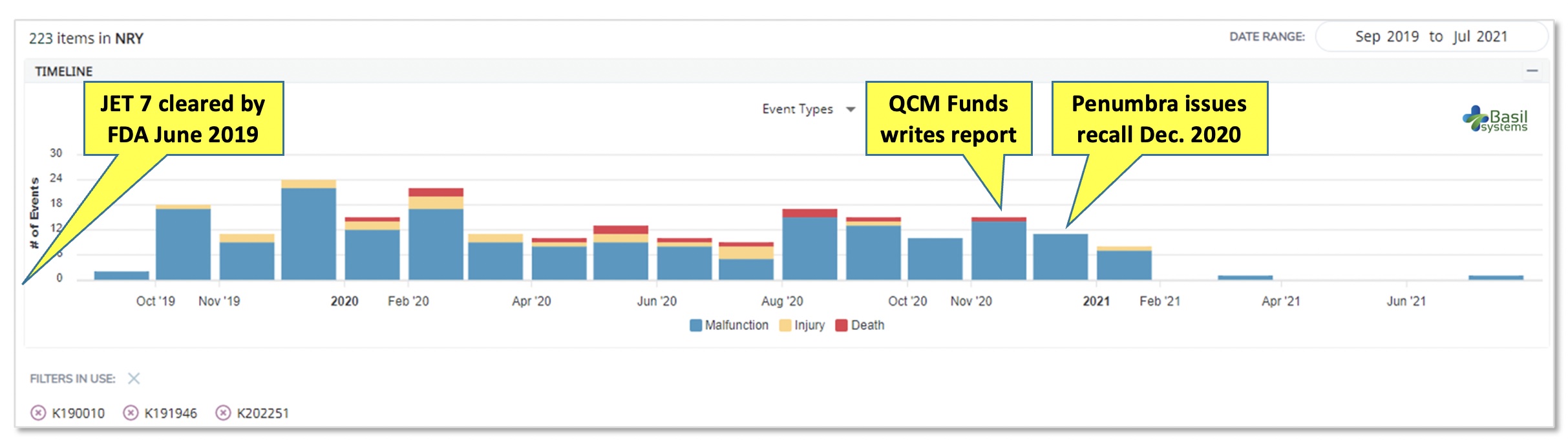 The data shows an immediate onset of reported AEs after commercial launch, but these did not attract much notice.
The data shows an immediate onset of reported AEs after commercial launch, but these did not attract much notice.
A year passed between the product’s market clearance and the recall. Adverse events – injuries first, then deaths – were reported within months. But, only after an activist investor, QCM Funds, published a report in late 2020 were the patient risks fully recognized and the product recalled from the market. Its withdrawal also triggered recalls for two related products with 510(k) pre-market submission applications: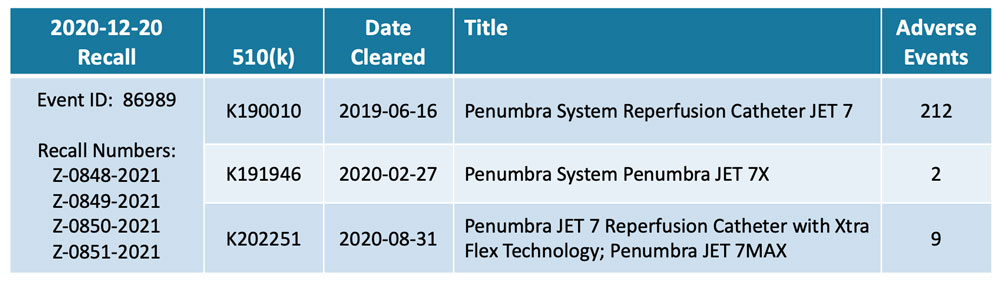
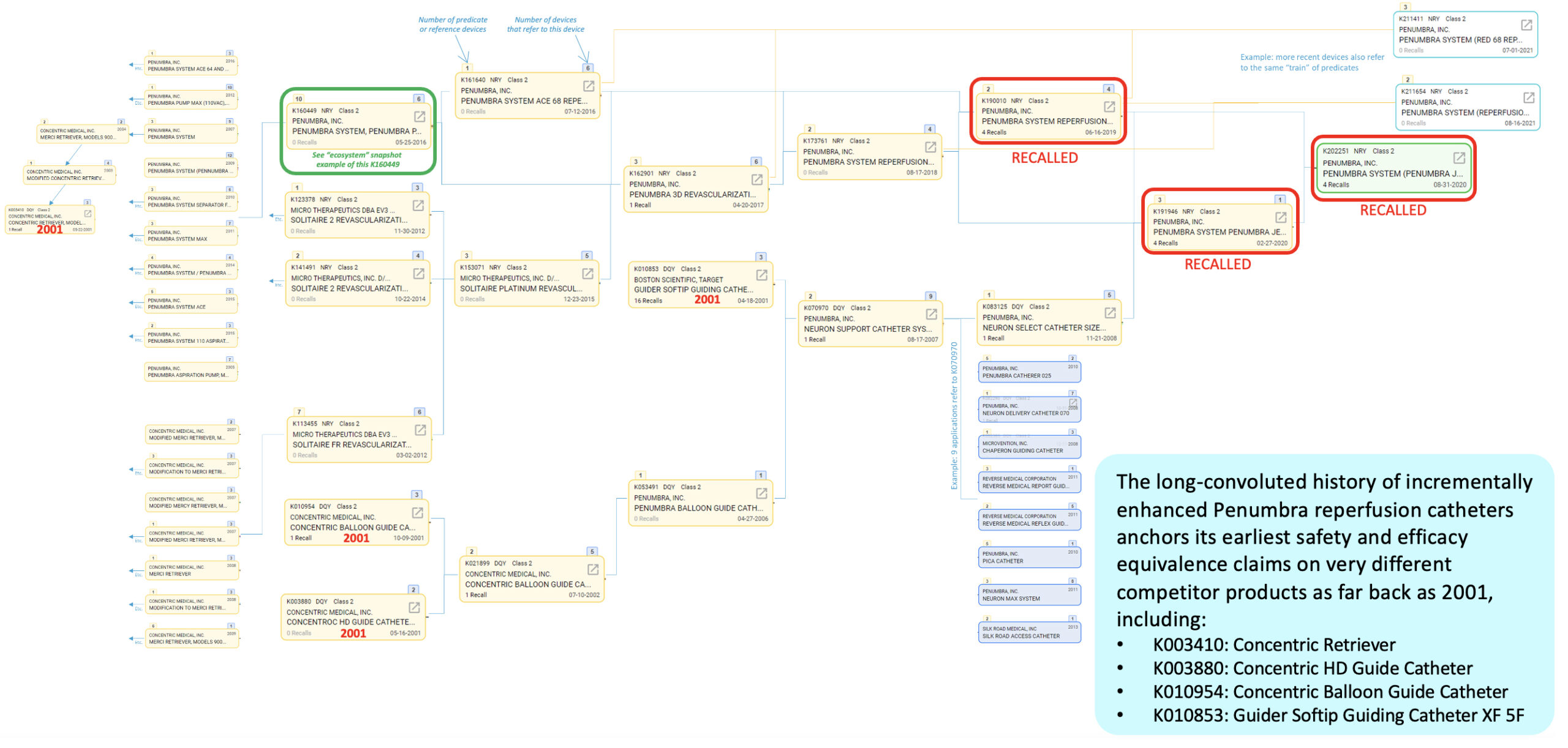
Interestingly, the long history of incrementally enhanced Penumbra reperfusion catheters anchors its earliest safety and efficacy equivalence claims on very different competitor products going as far back as 2001, such as:
- K003410: Concentric Retriever
- K003880: Concentric HD Guide Catheter
- K010954: Concentric Balloon Guide Catheter
- K010853: Guider Softip Guiding Catheter XF 5F
This is an example, and one that is not uncommon, of a historical weakness in the 510(k) clearance system, where the function of a modern device becomes difficult to connect to an early precedent’s function that may no longer closely resemble it.
Predicate history and “device creep” of JET 7’s reference devices
Our data also illustrates just how extensively some predicate devices are used across numerous newer devices. This may be appropriate it some cases, but it may also suggest that many different devices and functions have emerged and diverged from rather simple or basic early precedents.
A snapshot of one part of Penumbra’s predicate ecosystem
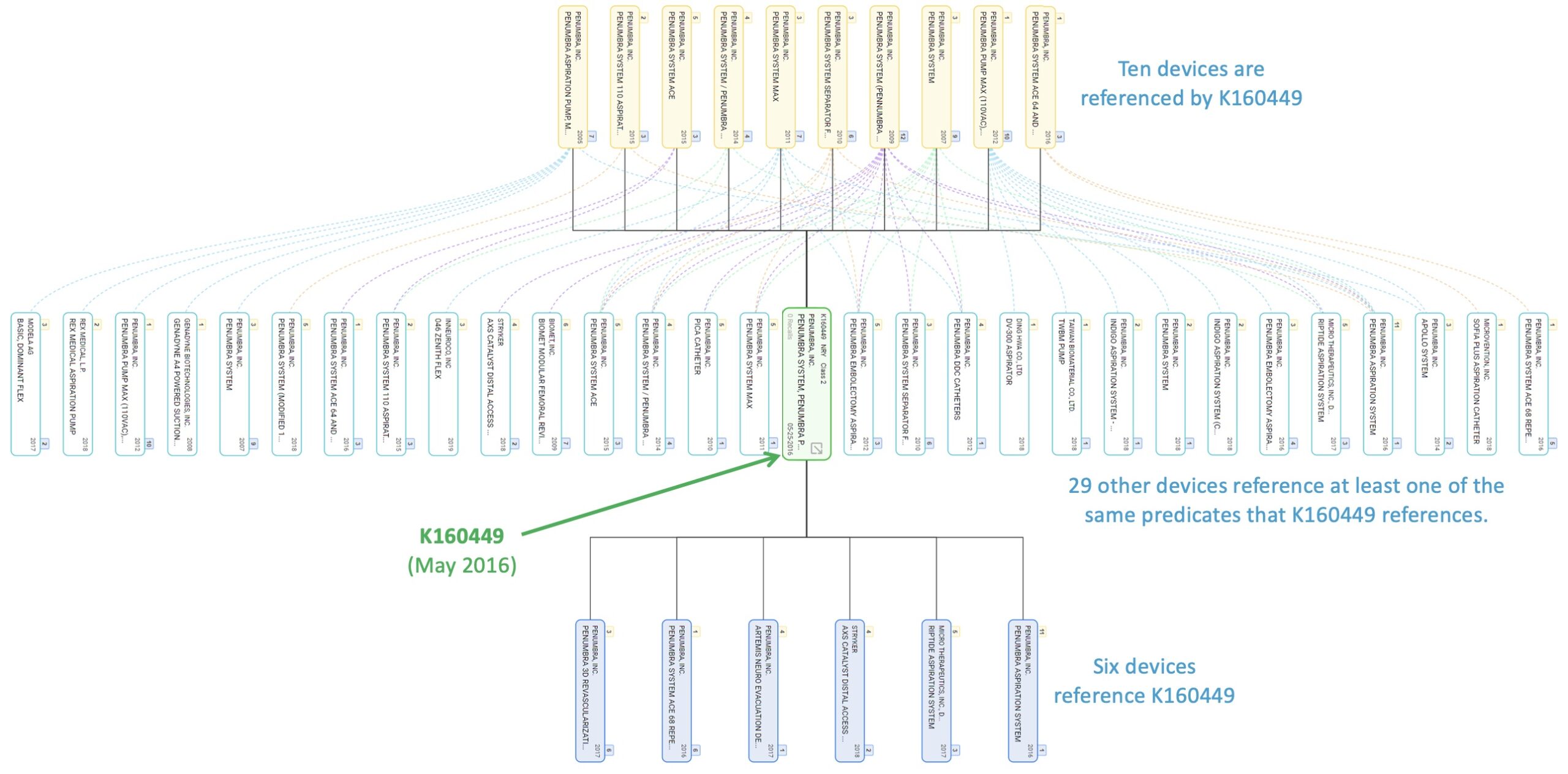
So, what does all this mean?
The data supports industry experts’ criticisms of the post-market surveillance system’s limitations and lack of oversight. It may also suggest a lack of responsiveness on the part of industry, which arguably could and should react to its emerging data and issue more timely recalls.
Navigate healthcare data with precision.
Basil Systems leverages big data and machine learning to provide healthcare enterprises with unprecedented analytics and insights that guide effective product and market strategies. Learn more about how we can help you reduce risks and launch faster.